So I’ve been fighting with this error for quite a few days and desperate. I’m currently working on a CNN with 70 images.
The size of X_train = 774164.
But, whenever I try to run this –> history = cnn.fit(X_train, y_train, batch_size=batch_size, epochs=epoch, validation_split=0.2) it leaks my memory and I get the next error:
W tensorflow/core/common_runtime/bfc_allocator.cc:457] Allocator (GPU_0_bfc) ran out of memory trying to allocate 5.04GiB (rounded to 5417907712)requested by op _EagerConst
If the cause is memory fragmentation maybe the environment variable ‘TF_GPU_ALLOCATOR=cuda_malloc_async’ will improve the situation.
I’m working with a bunch of guys that are developing a new platform for cloud GPU rental, and we’re still in the conception stages atm. I understand that not all of you are using GPU clouds, but for those of you who are, are there any features you think that current platforms are missing? Do you think there’s much room for improvement in the platforms you’ve used so far? What’s your favourite platform?
It would be great to get some insight from people who know what theyre talking about 🙂 TIA!
SANTA CLARA, Calif., Feb. 22, 2022 (GLOBE NEWSWIRE) — NVIDIA will present at the following event for the financial community: Morgan Stanley Technology, Media & Telecom ConferenceMonday, …
Clara Parabricks v3.7 supports gene panels, RNA-Seq, short tandem repeats, and updates to GATK (4.2) and DeepVariant (1.1), and PON support for Mutect2.
In the January 2022 release of Clara Parabricks v3.7, NVIDIA expanded the scope of the toolkit to new data types while continuing to improve upon existing tools:
Added support for analysis of RNASeq analysis
Added support for UMI-based gene panel analysis with an accelerated implementation of Fulcrum Genomics’ fgbio pipeline
Added support for mutect2 Panel of Normals (PON) filtering to bring the accelerated mutectcaller in line with the GATK best practices for calling tumor-normal samples
Incorporated a bam2fq method that enables accelerated realignment of reads to new references
Added support for short tandem repeat assays with ExpansionHunter
Accelerated post-calling VCF analysis steps by up to 15X
Updated HaplotypeCaller to match GATK v4.1 and updated DeepVariant to v1.1.
Clara Parabricks v3.7 significantly broadens the scope of what Clara Parabricks can do while continuing to invest in the field-leading whole genome and whole exome pipelines.
Enabling reference genome realignment with bam2fq and fq2bam
To address recent updates to the human reference genome and make realigning reads tractable for large studies, NVIDIA developed a new bam2fq tool. Parabricks bam2fq can extract reads in FASTQ format from BAM files, providing an accelerated replacement for tools like GATK SamToFastq or bazam.
Combined with Parabricks fq2bam, you can fully realign a 30X BAM file from one reference (for example, hg19) to an updated one (hg38 or CHM13) in 90 minutes using eight NVIDIA V100 GPUs. Internal benchmarks have shown that realigning to hg38 and rerunning variant calling captures several thousand more true-positive variants in the Genome in a Bottle HG002 truth set compared to relying solely on hg19.
The improvements in variant calls from realignment are almost the same as initially aligning to hg38. While this workflow was possible before, it was prohibitively slow. NVIDIA has finally made reference genome updates practical for even the largest of WGS studies in Clara Parabricks.
More options for RNASeq transcript quantification and fusion calling in Clara Parabricks
With version 3.7, Clara Parabricks adds two new tools for RNASeq analysis as well.
Transcript quantification is one of the most performed analyses for RNASeq data. Kallisto is a rapid method for expression quantification that relies on pseudoalignment. While Clara Parabricks already included STAR for RNASeq alignment, Kallisto adds a complementary method that can run even faster.
Fusion calling is another common RNASeq analysis. In Clara Parabricks 3.7, Arriba provides a second method for calling gene fusions based on the output of the STAR aligner. Arriba can call significantly more types of events than STAR-Fusion, including the following:
Viral integration sites
Internal tandem duplications
Whole exon duplications
Circular RNAs
Enhancer-hijacking events involving immunoglobulin and T-cell receptor loci
Breakpoints in introns and intergenic regions
Together, the addition of Kallisto and Arriba make Clara Parabricks a comprehensive toolkit for many transcriptome analyses.
Simplifying and accelerating gene panel and UMI analysis
While whole genome and whole exome sequencing are increasingly common in both research and clinical practice, gene panels dominate the clinical space.
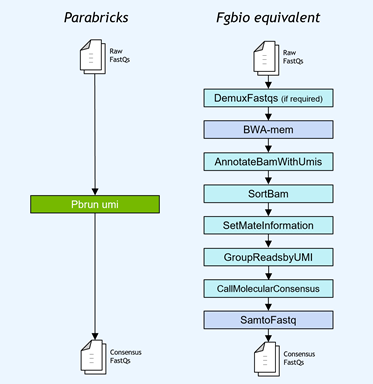
Gene panel workflows commonly use unique molecular identifiers (UMIs) attached to reads to improve the limits of detection for low-frequency mutations. NVIDIA accelerated the Fulcrum Genomics fgbio UMI pipeline and consolidated the eight-step pipeline into a single command in v3.7, with support for multiple UMI formats.
Figure 1. The Fulcrum Genomics Fgbio UMI pipeline accelerated with a single command on Clara Parabricks
Detecting changes in short tandem repeats with ExpansionHunter
Short tandem repeats (STRs) are well-established causes of certain neurological disorders as well as historically important markers for fingerprinting samples for forensic and population genetic purposes.
NVIDIA enabled genotyping of these sites in Clara Parabricks by adding support for ExpansionHunter in version 3.7. It’s now easy to go from raw reads to genotyped STRs entirely using the Clara Parabricks command-line interface.
Improving MuTect somatic mutation calls with PON support
It is common practice to filter somatic mutation calls against a set of mutations from known normal samples, also called a Panel of Normals (PON). NVIDIA added support for both publicly available PON sets and custom PONs to the mutectcaller tool, which now provides an accelerated version of the GATK best practices for somatic mutation calling.
Accelerating post-calling VCF annotation and quality control
In the v3.6 release, NVIDIA added the vbvm, vcfanno, frequencyfiltration, vcfqc, and vcfqcbybam tools that made post-calling VCF merging, annotation, filtering,filtering, and quality control easier to use.
The v3.7 release improved upon these tools by completely rewriting the backend of vbvm, vcfqc and vcfqcbybam, all of which are now more robust and up to 15x faster.
In the case of vcfanno, NVIDIA developed a new annotation tool called snpswift, which brings more functionality and acceleration while retaining the essential functionality of accurate allele-based database annotation of VCF files. The new snpswift tool also supports annotating a VCF file with gene name data from ENSEMBL, helping to make sense of coding variants. While the new post-calling pipeline looks similar to the one from v3.6, you should find that your analysis runs even faster.
With Clara Parabricks v3.7, NVIDIA is demonstrating a commitment to making Parabricks the most comprehensive solution for accelerated analysis of genomic data. It is an extensive toolkit for WGS, WES, and now RNASeq analysis, as well as gene panel and UMI data.
For more information about version 3.7, see the following resources:
Am I the only one that has webpage problems? Today I wanted to see some tutorials on their page and some links downloads xml files instead of showing the actual page.
With more than 11,000 stores across Thailand serving millions of customers, CP All, the country’s sole licensed operator of 7-Eleven convenience stores, recently turned to AI to dial up its call centers’ service capabilities. Built on the NVIDIA conversational AI platform, the Bangkok-based company’s customer service bots help call-center agents answer frequently asked questions and Read article >
This is, without a doubt, the best time to jump into cloud gaming. GeForce NOW RTX 3080 memberships deliver up to 1440p resolution at 120 frames per second on PC, 1600p and 120 FPS on Mac, and 4K HDR at 60 FPS on NVIDIA SHIELD TV, with ultra-low latency that rivals many local gaming experiences. Read article >
I am trying to adapt this to take in data that is divided into 1152 16bit little endian signed data;
The file shape is a 1D array.
The data is aligned (zero padded).
bash du -b *.raw 41575680 labels.raw 41575680 input.raw
Traceback: “`python Traceback (most recent call last): File “/home/test/./test.py”, line 22, in <module> model.fit(inputa, label, epochs=10) File “/usr/lib/python3.10/site-packages/keras/utils/traceback_utils.py”, line 67, in error_handler raise e.with_traceback(filtered_tb) from None File “/usr/lib/python3.10/site-packages/tensorflow/python/framework/func_graph.py”, line 1147, in autograph_handler raise e.ag_error_metadata.to_exception(e) ValueError: in user code:
File "/usr/lib/python3.10/site-packages/keras/engine/training.py", line 1021, in train_function * return step_function(self, iterator) File "/usr/lib/python3.10/site-packages/keras/engine/training.py", line 1010, in step_function ** outputs = model.distribute_strategy.run(run_step, args=(data,)) File "/usr/lib/python3.10/site-packages/keras/engine/training.py", line 1000, in run_step ** outputs = model.train_step(data) File "/usr/lib/python3.10/site-packages/keras/engine/training.py", line 859, in train_step y_pred = self(x, training=True) File "/usr/lib/python3.10/site-packages/keras/utils/traceback_utils.py", line 67, in error_handler raise e.with_traceback(filtered_tb) from None File "/usr/lib/python3.10/site-packages/keras/engine/input_spec.py", line 248, in assert_input_compatibility raise ValueError( ValueError: Exception encountered when calling layer "sequential" (type Sequential). Input 0 of layer "dense" is incompatible with the layer: expected axis -1 of input shape to have value 1152, but received input with shape (32, 1) Call arguments received: • inputs=tf.Tensor(shape=(32,), dtype=float32) • training=True • mask=None
“`
What’s the issue here? The data is 1152 samples/2304 bytes aligned.
How does it enter a blackhole and turn into (32,1)
” Input 0 of layer “dense” is incompatible with the layer: expected min_ndim=2, found ndim=1. Full shape received: (32,)” When I use InputLayer instead of Flatten.
I’ve been trying to get tensorflow to work with my rtx 3080 on anaconda without much success.
Im on windows and currently have toolkit 11.0 runtime libraries and cudnn 8.11 installed, which I use to remove stars in my astronomy images. I’m not sure how to get these to work in anaconda or how to get the other versions to work.

 Clara Parabricks v3.7 supports gene panels, RNA-Seq, short tandem repeats, and updates to GATK (4.2) and DeepVariant (1.1), and PON support for Mutect2.
Clara Parabricks v3.7 supports gene panels, RNA-Seq, short tandem repeats, and updates to GATK (4.2) and DeepVariant (1.1), and PON support for Mutect2.